ŌģĀ. ņä£ ļĪĀ
ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø(Cleidocranial dysplasia)ņØĆ ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ņØś ņ£ĀņĀäņ¦łĒÖśņ£╝ļĪ£, 2 - 3ņäĖ Ļ▓ĮņŚÉ ļéśĒāĆļéśļŖö ņćäĻ│© ļČĆņ£äņØś ļ¼┤ĒåĄņä▒ ņóģņ░ĮņØä ļ╣äļĪ»ĒĢśņŚ¼ ņćäĻ│©ņØś ĒśĢĒā£ ņØ┤ņāü, ļæÉĻ░£Ļ│© Ļ░ä ļ┤ēĒĢ® ņ¦ĆņŚ░, ņĀĆņŗĀņן, ņāüņĢģņØś ņŚ┤ņä▒ņן, Ļ│╝ņ×ēņ╣ś, ņśüĻĄ¼ņ╣ś ļ¦╣ņČ£ ņ¦ĆņŚ░ ļō▒ņØä ĒŖ╣ņ¦Ģņ£╝ļĪ£ ĒĢ£ļŗż. ņØ┤ ņ¦łĒÖśņØĆ 0.0001%ņØś ņ£Āļ│æļźĀņØä ļ│┤ņØ┤ļ®░, ĒÖśņ×ÉņØś 60 - 70%ņŚÉņä£ Runt-related transcription factor 2 (RUNX2) ņ£ĀņĀäņ×ÉņØś Ļ▓░ĒĢ©ņØ┤ ĒÖĢņØĖļÉ£ļŗż[1-3].
RUNX2ļŖö 6ļ▓ł ņŚ╝ņāēņ▓┤ņŚÉ ņĪ┤ņ×¼ĒĢśļŖö 8Ļ░£ņØś exonņ£╝ļĪ£ ņØ┤ļŻ©ņ¢┤ņ¦ä ņ£ĀņĀäņ×ÉļĪ£ņä£, Core Binding Factor A1 (CBFA1)ņØ┤ļØ╝Ļ│ĀļÅä ĒĢ£ļŗż. RUNX2 ļŗ©ļ░▒ņ¦łņØĆ ļŗżņ¢æĒĢ£ ĻĖ░ļŖźņä▒ ļÅäļ®öņØĖ(functional domain)ņØä ĒżĒĢ©ĒĢśļŖöļŹ░, ņØ┤ ņżæ 2 - 4 exonņŚÉ Ļ▒Ėņ│É ņĪ┤ņ×¼ĒĢśļŖö RHD (Runt-DNA binding homology domain)ļŖö ļ¬©ļōĀ Runt ņ£ĀņĀäņ×ÉņĪ▒(gene family)ņŚÉ Ļ│ĄĒåĄņĀüņ£╝ļĪ£ ļ│┤ņĪ┤ļÉśņ¢┤ ņ׳ļŖö ņżæņÜöĒĢ£ ļČĆņ£äņØ┤ļŗż. RHDļŖö 128Ļ░£ņØś ņĢäļ»ĖļģĖņé░ņ£╝ļĪ£ ĻĄ¼ņä▒ļÉśļ®░, RUNX2Ļ░Ć DNAņŚÉ Ļ▓░ĒĢ®ĒĢśļŖö Ļ▓āņØä ļÅäņÖĆ ļŗ©ļ░▒ņ¦łņØś ņ×æņÜ®ņŚÉ ĒĢäņłśņĀüņØ┤ļŗż[4].
RUNX2ļŖö RHD ļČĆņ£äļź╝ ĒåĄĒĢ┤ Ļ▓ĮņĪ░ņ¦ü ĒśĢņä▒ Ļ┤ĆļĀ© ņ£ĀņĀäņ×ÉņØś ĒĢĄņŗ¼Ļ▓░ĒĢ® ņØĖņ×É ļČĆņ£ä(osteoblast-specific cis-acting element 2, OSE2)ņŚÉ Ļ▓░ĒĢ®ĒĢśņŚ¼ Ēæ£ņĀü ņ£ĀņĀäņ×ÉņØś ļ░£ĒśäņØä ņĪ░ņĀłĒĢ£ļŗż[5]. OSE2ļŖö OCN (osteocalcin), IĒśĢ COL (collagen), BSP (bone sialoprotein), OPN (osteopontin), MMP-13 (matrix metalloproteinase 13) ļō▒ņØś ļŗżņ¢æĒĢ£ Ļ│© ĻĖ░ņ¦ł ļŗ©ļ░▒ņ¦ł(bone matrix protein)ņØś ĒöäļĪ£ļ¬©Ēä░ ņśüņŚŁņŚÉņä£ ļ░£Ļ▓¼ļÉśĻ│Ā, ņ╣śņĢäņŚÉ ĒŖ╣ņ¦ĢņĀüņØĖ ĻĖ░ņ¦ł ļŗ©ļ░▒ņ¦łņØĖ AMBN (ameloblastin)ņØś ĒöäļĪ£ļ¬©Ēä░ ņśüņŚŁņŚÉņä£ļÅä ĒÖĢņØĖļÉ£ļŗż[6-8].
ļśÉĒĢ£, RUNX2ļŖö Ļ░äņŚĮņĀäĻĄ¼ņäĖĒż(mesenchymal progenitor cell)Ļ░Ć ņĪ░Ļ│©ņäĖĒż(osteoblast)ļĪ£ ļČäĒÖöĒĢśļŖö Ļ│╝ņĀĢņØś Ļ░ü ļŗ©Ļ│äņŚÉ Ļ┤ĆņŚ¼ĒĢśĻ│Ā, ļ╣äļīĆņŚ░Ļ│©ņäĖĒż(hypertrophic chondrocyte)ņØś ĒśĢņä▒ņØä ņ£ĀļÅäĒĢśņŚ¼ Ļ│©ņØś ĒśĢņä▒ņŚÉļÅä ņ¦üņĀæņĀüņ£╝ļĪ£ ņ×æņÜ®ĒĢ£ļŗż[6,9]. ļö░ļØ╝ņä£ RUNX2ņØś ņØ┤ņāüņØĆ ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”ØņŚÉņä£ ļéśĒāĆļéśļŖö ļŗżņ¢æĒĢ£ Ēæ£ĒśäĒśĢņØś ņøÉņØĖņ£╝ļĪ£ ņ×æņÜ®ĒĢĀ ņłś ņ׳ļŗż.
RUNX2ļŖö Ēśäņ×¼Ļ╣īņ¦Ć ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ ņĢīļĀżņ¦ä ņ£ĀņØ╝ĒĢ£ ņøÉņØĖ ņ£ĀņĀäņ×ÉļĪ£, ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ ļ│┤Ļ│ĀļÉ£ RUNX2ņØś ļÅīņŚ░ļ│ĆņØ┤ļŖö 100Ļ░Ćņ¦Ć ņØ┤ņāüņ£╝ļĪ£ ļŗżņ¢æĒĢśļŗż. ĒĢśņ¦Ćļ¦ī RUNX2ņØś ĒÖ£ņä▒ņØ┤ ļŗżņ¢æĒĢ£ ļÅäļ®öņØĖņØś ņ×æņÜ®Ļ│╝ ņĀäņé¼ Ēøä ļ│ĆĒśĢ(posttranscriptional modification) ļō▒ņŚÉ ņØśĒĢ┤ ņĪ░ņĀłļÉśņ¢┤ Ēæ£ĒśäĒśĢņ£╝ļĪ£ ļéśĒāĆļéśĻĖ░ ļĢīļ¼ĖņŚÉ ņ¦üņĀæņĀüņØĖ ņ£ĀņĀäņ×ÉĒśĢ-Ēæ£ĒśäĒśĢ ņāüĻ┤Ć Ļ┤ĆĻ│ä(genotype-phenotype correlation)ļź╝ ĒÖĢļ”ĮĒĢśļŖö Ļ▓āņØĆ ņēĮņ¦Ć ņĢŖļŗż[10].
ņØ┤ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ĒŖ╣ņ¦ĢņĀüņØĖ ĻĄ¼Ļ░Ģ ļé┤ ņåīĻ▓¼ņØä Ļ░Ćņ¦ĆļŖö ĒĢ£ĻĄŁņØĖ ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ RUNX2 ņ£ĀņĀäņ×ÉņØś ņŚ╝ĻĖ░ ņä£ņŚ┤ ļČäņäØņØä ņŗ£Ē¢ēĒĢśņŚ¼ ļ│æņØĖņØä ĻĘ£ļ¬ģĒĢśĻ│Ā, ĻĘĖ ĻĖ░ļŖźņŚÉ ļīĆĒĢ┤ Ļ│Āņ░░ĒĢśĻ│Āņ×É ĒĢśņśĆļŗż.
ŌģĪ. ņŚ░ĻĄ¼ ņ×¼ļŻī ļ░Å ļ░®ļ▓Ģ
1. Ēæ£ļ│Ė
ļŗżņłśņØś ņśüĻĄ¼ņ╣ś ļ¦╣ņČ£ ņ¦ĆņŚ░ņØä ņŻ╝ņåīļĪ£ ļŗ©ĻĄŁļīĆĒĢÖĻĄÉ ņ╣śĻ│╝ļ│æņøÉ ņåīņĢäņ╣śĻ│╝ņŚÉņä£ ņ╣śļŻīļź╝ ļ░øņĢäņś© 23ņäĖ ņŚ¼ĒÖśņ£╝ļĪ£ņä£, ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Øņ£╝ļĪ£ ņØśņŗ¼ļÉśņ¢┤ ņä£ņÜĖļīĆĒĢÖĻĄÉ ņ╣śĻ│╝ļ│æņøÉ ņåīņĢäņ╣śĻ│╝ņŚÉ ņ£ĀņĀäĻ▓Ćņé¼ļź╝ ņ£äĒĢ┤ņä£ ņØśļó░ļÉśņŚłļŗż. ĒÖśņ×ÉņÖĆ ĒÖśņ×ÉņØś ņ¢┤ļ©Ėļŗłļź╝ ļīĆņāüņ£╝ļĪ£ ņŚ░ĻĄ¼ ņ░ĖņŚ¼ ļÅÖņØśņä£ļź╝ ļ░øņØĆ Ēøä, ĻĄ¼Ļ░Ģ Ļ▓Ćņé¼ ļ░Å ļ░®ņé¼ņäĀ ņé¼ņ¦ä ņ┤¼ņśüņØä ņ¦äĒ¢ēĒĢśņśĆĻ│Ā, ļ¦Éņ┤ł ĒśłņĢĪņØä ņ▒äņĘ©ĒĢśņśĆļŗż. ĒĢ┤ļŗ╣ ņŚ░ĻĄ¼ ĒöäļĪ£ĒåĀņĮ£ņØĆ ņä£ņÜĖļīĆĒĢÖĻĄÉņ╣śĻ│╝ļ│æņøÉ Institutional Review BoardņØś ņŖ╣ņØĖ ņĢäļל ņ¦äĒ¢ēĒĢśņśĆļŗż(IRB File NO. : GRI05003G).
2. Primer ņĀ£ņ×æ ļ░Å ņżæĒĢ®ĒÜ©ņåī ņŚ░ņćä ļ░śņØæ(Polymerase chain reaction, PCR)
QuickGene DNA whole blood kit SņÖĆ QuickGene-Mini80 equipment (Fujifilm, Tokyo, Japan)ļź╝ ņØ┤ņÜ®ĒĢśņŚ¼, ņ▒äņĘ©ĒĢ£ ļ¦Éņ┤ł ĒśłņĢĪņŚÉņä£ genomic DNAļź╝ ņČöņČ£ĒĢśņśĆļŗż. RUNX2ņØś 8Ļ░£ exon ņśüņŚŁņØä ņ”ØĒÅŁĒĢśĻĖ░ ņ£äĒĢśņŚ¼ ņé¼ņÜ®ĒĢ£ primerļŖö ļŗżņØīĻ│╝ Ļ░Öļŗż(Table 1). ņżæĒĢ®ĒÜ©ņåī ņŚ░ņćä ļ░śņØæņØĆ HiPi DNA polymerase premix (ElpisBio, Daejeon, Korea)ļź╝ ņØ┤ņÜ®ĒĢśņŚ¼ ņ¦äĒ¢ēĒĢśņśĆĻ│Ā, ņ”ØĒÅŁļÉ£ ņé░ļ¼╝ņØĆ PCR Purification kit (ElpisBio, Daejeon, Korea)ļź╝ ņé¼ņÜ®ĒĢśņŚ¼ ņĀĢņĀ£ĒĢśņśĆļŗż.
3. DNA ņŚ╝ĻĖ░ ņä£ņŚ┤ ļČäņäØ
ņĀĢņĀ£ļÉ£ PCR ņé░ļ¼╝ņØĆ DNA sequencing center (Macrogen, Seoul, Korea)ņŚÉņä£ DNA ņŚ╝ĻĖ░ ņä£ņŚ┤ ļČäņäØņØä ņŗ£Ē¢ēĒĢśņśĆļŗż. Ļ▓░Ļ│╝ļŖö NCBI GeneBankņØś ņ░ĖĻ│Ā ņä£ņŚ┤Ļ│╝ ļ╣äĻĄÉĒĢśņŚ¼ ĒĢ┤ļŗ╣ ņśüņŚŁņØś ļÅīņŚ░ļ│ĆņØ┤ļź╝ ņĪ░ņé¼ĒĢśņśĆļŗż. ļ¬©ļōĀ ĒĢĄņé░(nucleotide)ņØś ļ▓łĒśĖļŖö RUNX2 ļÅÖĒśĢ mRNA ņżæ NM_001024630.4 (isoform a)ļź╝ ĻĖ░ņżĆņ£╝ļĪ£ ĒĢśņśĆļŗż.
Ōģó. ņŚ░ĻĄ¼ ņä▒ņĀü
1. ņ×äņāüņĀü ļ░Å ļ░®ņé¼ņäĀĒĢÖņĀü Ļ▓Ćņé¼ Ļ▓░Ļ│╝
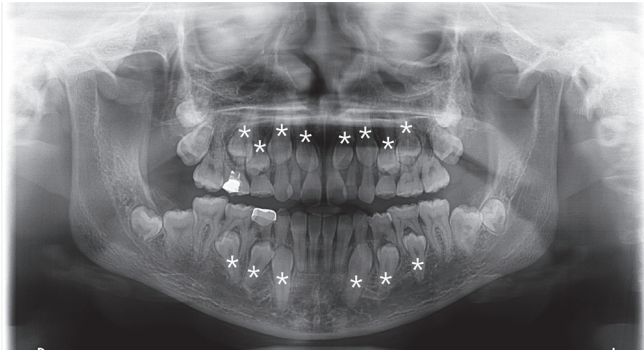
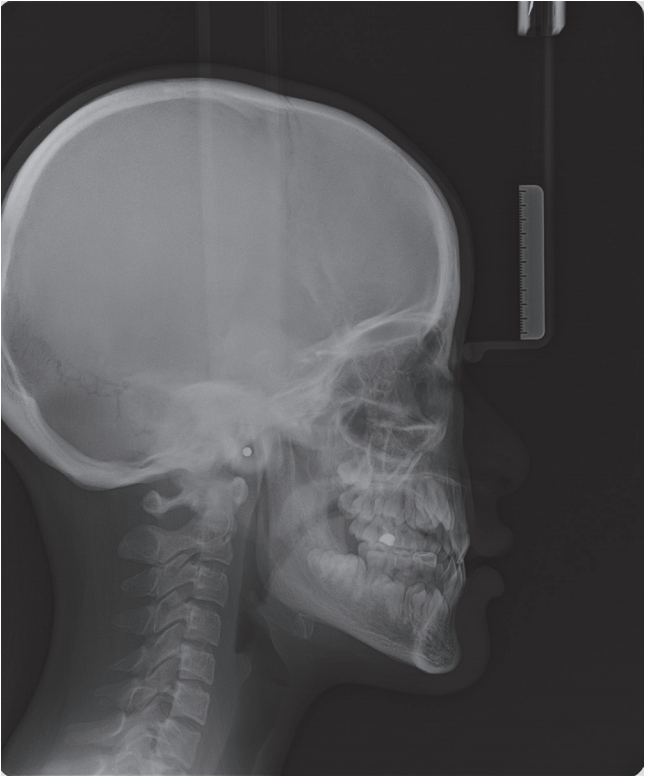
ĒÖśņ×ÉļŖö ņ×äņāüņĀü Ļ▓Ćņé¼ Ļ▓░Ļ│╝ ĻĄ¼Ļ░Ģ ļé┤ņŚÉ ļŗżņłśņØś ņ£Āņ╣śĻ░Ć ņ×öņĪ┤ĒĢ┤ ņ׳ņŚłļŗż. ĒÖśņ×ÉņØś ļ░®ņé¼ņäĀ ņé¼ņ¦äņŚÉņä£ļŖö ņāüņĢģ ņĖĪņĀłņ╣ś, Ļ▓¼ņ╣ś, ņĀ£1ņåīĻĄ¼ņ╣ś, ņĀ£2ņåīĻĄ¼ņ╣ś ļ░Å ĒĢśņĢģ Ļ▓¼ņ╣ś, ņĀ£1ņåīĻĄ¼ņ╣ś, ņĀ£2ņåīĻĄ¼ņ╣ś ļō▒ ļŗżņłś ņ╣śņĢäņØś ļ¦╣ņČ£ ņ¦ĆņŚ░ņØ┤ Ļ┤Ćņ░░ļÉśņŚłĻ│Ā, ļæÉĻ░£Ļ│© ļ┤ēĒĢ® ņ¦ĆņŚ░ ļśÉĒĢ£ Ļ┤Ćņ░░ļÉśņŚłļŗż(Fig. 1, 2, Table 1). ĒÖśņ×É ņ¢┤ļ©ĖļŗłņØś ļ░®ņé¼ņäĀ ņé¼ņ¦äņŚÉņä£ļŖö ĒĢśņĢģ ņóīņĖĪ Ļ▓¼ņ╣śņØś ļ¦żļ│ĄņØ┤ ĒÖĢņØĖļÉśņŚłļŗż(Fig. 3). ĒÖśņ×ÉņÖĆ ņ¢┤ļ©Ėļŗł ļ¬©ļæÉņŚÉņä£ Ļ│╝ņ×ēņ╣śļŖö Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖņĢśĻ│Ā(Fig. 1, 3), Ļ│╝ņ×ēņ╣ś ļ░£Ļ▒░ ļ│æļĀźļÅä ĒÖĢņØĖļÉśņ¦Ć ņĢŖņĢśļŗż.
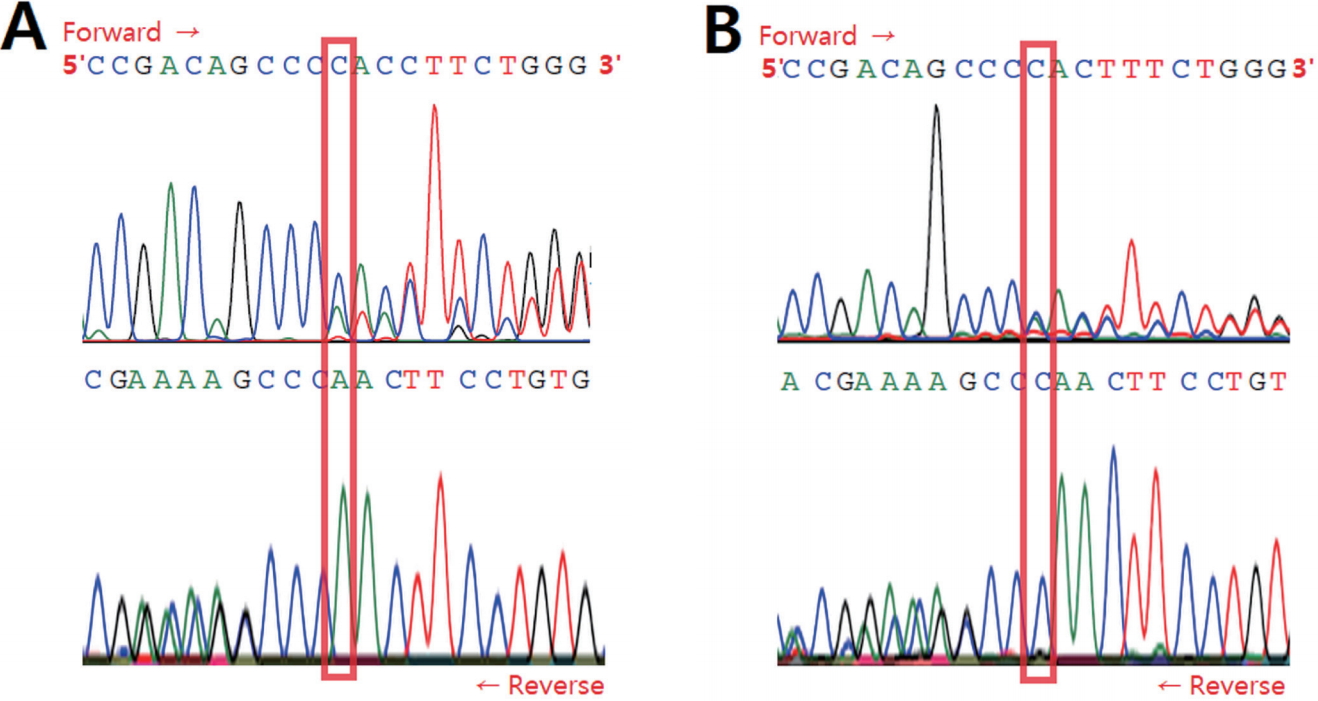
2. ņŚ╝ĻĖ░ ņä£ņŚ┤ ļČäņäØ Ļ▓░Ļ│╝
RUNX2 ņ£ĀņĀäņ×ÉņØś ņŚ╝ĻĖ░ ņä£ņŚ┤ ļČäņäØ Ļ▓░Ļ│╝, ļīĆņāüņ×É ļ¬©ļæÉņŚÉņä£ ņØ┤ņĀäņŚÉ ļ│┤Ļ│ĀļÉśņ¦Ć ņĢŖņØĆ ņāłļĪ£ņÜ┤ ļ│ĆņØ┤ļź╝ ĒÖĢņØĖĒĢśņśĆļŗż. 3ļ▓ł exon ļé┤ņØś 357ļ▓ł ņŚ╝ĻĖ░ņä£ņŚ┤ņØĖ ņŗ£ĒåĀņŗĀ(cytosine)ņØ┤ ņĀ£Ļ▒░ļÉ£ ļ│ĆņØ┤Ļ░Ć ĒÖĢņØĖļÉśņŚłĻ│Ā(NM_001024630.4: c.357delC), ņØ┤ļŖö 2Ļ░£ņØś ļīĆļ”Įņ£ĀņĀäņ×É(allele) ņżæ ĒĢ£ ņ¬ĮņŚÉņä£ļ¦ī ļ░£Ļ▓¼ļÉ£ ņØ┤ĒśĢņĀæĒĢ®(heterozygous)ņ£╝ļĪ£ ļéśĒāĆļé¼ļŗż(Fig. 4). ļ░£Ļ▓¼ļÉ£ ļÅīņŚ░ļ│ĆņØ┤ļĪ£ ņØĖĒĢ┤ ĒĢ┤ļŗ╣ ņśüņŚŁņŚÉņä£ ļ▓łņŚŁļÉśļŖö ņĢäļ»ĖļģĖņé░ņØ┤ ņĢäņŖżĒīīļØ╝ņ¦ä(asparagine)ņŚÉņä£ ĒŖĖļĀłņśżļŗī(threonine)ņ£╝ļĪ£ ņ╣śĒÖśļÉśĻ│Ā, ņØ┤ĒøäņØś ņŚ╝ĻĖ░ņä£ņŚ┤ņØś -1 net frameshiftļĪ£ ņØĖĒĢ┤ ļÅīņŚ░ļ│ĆņØ┤ ņ£äņ╣śņŚÉņä£ļČĆĒä░ 24ļ▓łņ¦Ė ņĢäļ»ĖļģĖņé░ņØ┤ ņóģĻ▓░ņĮöļÅłņ£╝ļĪ£ ņ╣śĒÖśļÉ£ļŗż[p.(Asn120Thrfs*24)].
ŌģŻ. ņ┤ØĻ┤ä ļ░Å Ļ│Āņ░░
ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”ØņØĆ RUNX2ņØś Ļ▓░ĒĢ© ļō▒ņ£╝ļĪ£ ņØĖĒĢ┤ ļ░£ņāØĒĢśļŖö ņ¦łĒÖśņ£╝ļĪ£, ļŗżņ¢æĒĢ£ Ļ│©Ļ▓®ņä▒ ņØ┤ņāüņØä ļÅÖļ░śĒĢśņ¦Ćļ¦ī, ņØ╝ņāü ņāØĒÖ£ņØä ĒĢśļŖöļŹ░ Ēü░ ņןņĢĀļź╝ ņ£Āļ░£ĒĢśņ¦Ć ņĢŖļŖö Ļ▓ĮņÜ░Ļ░Ć ļ¦ÄņĢä, ņ╣śņä▒ ļ¼ĖņĀ£ņØś ļ░£Ļ▓¼ņØ┤ ņ¦łļ│æņØś ņ¦äļŗ©Ļ│╝ņĀĢņŚÉ ņżæņÜöĒĢ£ ļŗ©ņä£Ļ░Ć ļÉ£ļŗż.
ņ╣śņĢä ļ¦╣ņČ£ ņ¦ĆņŚ░ņØĆ ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ ĒØöĒĢśĻ▓ī Ļ┤Ćņ░░ļÉśļŖö ĻĄ¼Ļ░Ģ ļé┤ ņ”ØņāüņØ┤ļŗż. ņ╣śņĢäņØś ļ¦╣ņČ£ņØĆ ņ╣śņĪ░Ļ│©ņØś ĒØĪņłśņÖĆ ņłśņ¦üņĀü ņ╣śņĢä ņØ┤ļÅÖņŚÉ ņØśĒĢ┤ ņØ┤ļŻ©ņ¢┤ņ¦ĆļŖöļŹ░, ņ╣śņĪ░Ļ│©ņØś ĒØĪņłśļź╝ ĒåĄĒĢ£ ļ¦╣ņČ£ļĪ£ņØś ĒśĢņä▒ņØĆ ņĪ░Ļ│©ņäĖĒżņÖĆ ĒīīĻ│©ņäĖĒżņØś ņāüĒśĖņ×æņÜ®ņŚÉ ņØśĒĢ┤ ņĪ░ņĀłļÉ£ļŗż. ĒīīĻ│©ņäĖĒżņØś ĒśĢņä▒ņØĆ RANK-RANKL (receptor activator of nuclear factor-╬║B ligand) ņŗĀĒśĖņĀäļŗ¼ņ▓┤Ļ│äņŚÉ ņØśĒĢ┤ ņ£ĀļÅäļÉśĻ│Ā, OPG (osteoprotegerin)ņŚÉ ņØśĒĢ┤ ņ¢ĄņĀ£ļÉ£ļŗż[11]. RANKL ļ░Å OPGņØś ĒöäļĪ£ļ¬©Ēä░ ņśüņŚŁņŚÉņä£ RUNX2ņØś Ļ▓░ĒĢ® ļČĆņ£äĻ░Ć ĒÖĢņØĖļÉśņŚłņ£╝ļ®░, ņ╣śņĢä ļ¦╣ņČ£ ņŗ£ĻĖ░ņŚÉ RUNX2Ļ░Ć ņĀüņĀłĒĢśĻ▓ī ļ░£ĒśäļÉśņ¦Ć ļ¬╗ĒĢśļ®┤ ĒīīĻ│©ņäĖĒżņØś ĒśĢņä▒ņŚÉ Ļ┤ĆņŚ¼ĒĢśļŖö ņØĖņ×ÉļōżņØ┤ ņČ®ļČäĒ׳ ĻĖ░ļŖźĒĢśņ¦Ć ļ¬╗ĒĢ┤ ļ¦╣ņČ£ Ļ▓ĮļĪ£ ņāüņØś ĒīīĻ│©ņäĖĒżĻ░Ć ļČĆņĪ▒ĒĢ┤ņ¦äļŗż. ņØ┤ļĪ£ ņØĖĒĢ┤ ņ╣śņĪ░Ļ│© ĒØĪņłśĻ░Ć ņØ┤ļŻ©ņ¢┤ņ¦Ćņ¦Ć ļ¬╗ĒĢśĻ│Ā ņ╣śņĢä ļ¦╣ņČ£ ņ¦ĆņŚ░ņØ┤ ņ£Āļ░£ļÉĀ ņłś ņ׳ļŗż[12-14].
Yoda ļō▒[15]ņØĆ Runx2 Ļ▓░ņåÉ ņØ┤ĒśĢ ņĀæĒĢ® ņźÉ(knock-out heterozygous mice)ļź╝ ņØ┤ņÜ®ĒĢśņŚ¼ ņ╣śņĢäņØś ĒśĢņä▒Ļ│╝ ļ¦╣ņČ£ņŚÉ ļīĆĒĢ£ Runx2ņØś ņśüĒ¢źņØä Ļ┤Ćņ░░ĒĢśņśĆļŗż. ņĀĢņāüĻĄ░Ļ│╝ ļ╣äĻĄÉ ņŗ£, ņāØĒøä 8 - 10ņØ╝ņŚÉ ņŗżĒŚśĻĄ░ņØś ņ╣śņĢä ĒśĢņä▒ņŚÉļŖö ļ¼ĖņĀ£Ļ░Ć ņŚåņŚłņ¦Ćļ¦ī, ņ╣śņĢä ļ¦╣ņČ£ ņ¦ĆņŚ░ņØ┤ Ļ┤Ćņ░░ļÉśņŚłļŗż. ņŗżĒŚśĻĄ░ņŚÉņä£ļÅä ņ╣śņĢä ļ¦╣ņČ£ Ļ▓ĮļĪ£ņāüņŚÉ ĒīīĻ│©ņäĖĒżņØś ņ”ØĻ░ĆĻ░Ć Ļ┤Ćņ░░ļÉśņŚłņ£╝ļéś, ņ╣śņĪ░Ļ│© ĒØĪņłśņÖĆ ņ╣śņĢä ļ¦╣ņČ£ņØä ņ£ĀļÅäĒĢśĻĖ░ņŚÉļŖö ņĀĢņāüĻĄ░ņŚÉ ļ╣äĒĢ┤ ĒīīĻ│©ņäĖĒżņØś ņ¢æņØ┤ ļČĆņĪ▒ĒĢśņŚ¼ ņ╣śņĢä ļ¦╣ņČ£ņØ┤ ļŗżņåī ļŖ”Ļ▓ī ņ¦äĒ¢ēļÉśļŖö Ļ▓āņØ┤ ĒÖĢņØĖļÉśņŚłļŗż. ņ£äņØś ņŚ░ĻĄ¼ļź╝ ĒåĄĒĢ┤, ņĀĢņāüņĀüņØĖ ņ╣śņĢä ļ¦╣ņČ£ņØä ņ£äĒĢ┤ RUNX2Ļ░Ć ņĀüņĀłĒĢ£ ņŗ£ĻĖ░ņŚÉ, ņĀüĒĢ®ĒĢ£ ņ£äņ╣śņŚÉņä£ ņČ®ļČäĒ׳ ļ░£ĒśäļÉśņ¢┤ņĢ╝ ĒĢ©ņØä ĒÖĢņØĖĒĢĀ ņłś ņ׳ļŗż.
ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ ņ╣śņĢä ļ¦╣ņČ£ ņ¦ĆņŚ░ņØĆ ņ£Āņ╣śņŚÉņä£ļŖö Ļ▒░ņØś ļ│┤Ļ│ĀļÉśņ¦Ć ņĢŖņĢśĻ│Ā, ņśüĻĄ¼ņ╣śņŚÉ ļīĆĒĢ┤ņä£ļÅä ņĀäņ╣śļČĆ ļ░Å ņĀ£1ļīĆĻĄ¼ņ╣śļ│┤ļŗżļŖö ņŻ╝ļĪ£ ņåīĻĄ¼ņ╣ś ļČĆņ£äņŚÉņä£ Ļ┤Ćņ░░ļÉśņŚłļŗż[16]. ņØ┤ļŖö ņāüņĢģĻ│©ņØś ĒøäĒć┤, ņĀäļæÉļČĆ ļÅīņČ£Ļ│╝ Ļ░ÖņØĆ ņĢģņĢłļ®┤ Ļ│©Ļ▓®ņØś ņØ┤ņāüņØ┤ ņåīņĢäņ▓ŁņåīļģäĻĖ░ ĒøäĻĖ░ņŚÉ ļæÉļō£ļ¤¼ņ¦ĆļŖö Ļ▓āĻ│╝ Ļ░ÖņØĆ ļ¦źļØĮņ£╝ļĪ£, RUNX2ņØś ņČ®ļČäĒĢ£ ļ░£ĒśäņØ┤ ņČ£ņāØ Ēøä ļ░£ļŗ¼ ĒøäĻĖ░ņŚÉ ņżæņÜöĒĢ£ Ļ▓āņ£╝ļĪ£ ņØ┤ĒĢ┤ĒĢĀ ņłś ņ׳ļŗż[17]. ĒĢśņ¦Ćļ¦ī ņØ┤ļ¤¼ĒĢ£ ņŗ£ĻĖ░ņĀü ĒŖ╣ņØ┤ņä▒ņØś ņĀĢĒÖĢĒĢ£ ņøÉņØĖņŚÉ ļīĆĒĢ┤ņä£ļŖö ņČöĻ░ĆņĀüņØĖ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśļŗż.
ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ ļéśĒāĆļéśļŖö ļśÉ ļŗżļźĖ ĻĄ¼Ļ░Ģ ļé┤ ĒŖ╣ņ¦ĢņĀüņØĖ ņ”Øņāüņ£╝ļĪ£ Ļ│╝ņ×ēņ╣śĻ░Ć ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż. ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ ļéśĒāĆļéśļŖö Ļ│╝ņ×ēņ╣śļŖö ņĀĢņāü ņ╣śņĢäņÖĆ ņ£Āņé¼ĒĢ£ ĒśĢĒā£ļź╝ ļØĀĻ│Ā, ņĀĢņāü ņśüĻĄ¼ņ╣śļ│┤ļŗż ļŗżņåī ļŖ”Ļ▓ī ļ░£ļŗ¼ĒĢśņŚ¼ ņ£Āņ╣śņŚ┤, ņśüĻĄ¼ņ╣śņŚ┤ņŚÉ ņØ┤ņØĆ ņĀ£3ņØś ņ╣śņŚ┤ ĒśĢĒā£ļĪ£ ņä▒ņןĒĢśĻ▓ī ļÉ£ļŗż[18].
Runx2Ļ░Ć Ļ▓░ņåÉļÉ£ ņźÉņŚÉņä£ ņ╣śņĢäņØś ĒśĢņä▒ ĒøäņŚÉļÅä ņāüĒö╝ ļÅīĻĖ░(epithelial bud)Ļ░Ć Ēć┤ĒÖöĒĢśņ¦Ć ņĢŖĻ│Ā ļÜ£ļĀĘĒĢśĻ▓ī Ļ┤Ćņ░░ļÉśņŚłņ£╝ļ®░, ņāüĒö╝ ļÅīĻĖ░ņŚÉņä£ Shh (Sonic hedgehog)Ļ░Ć Ļ░ĢĒĢśĻ▓ī ļ░£ĒśäļÉśļŖö Ļ▓āņØä ĒÖĢņØĖĒĢĀ ņłś ņ׳ņŚłļŗż[19]. ShhļŖö ņ╣śņĢäņØś ļ░£ļŗ¼ Ļ│╝ņĀĢņŚÉņä£ ĻĄ¼Ļ░Ģ ņāüĒö╝ņÖĆ ņ╣śņä▒ Ļ░äņŚĮņØś ņāüĒśĖņ×æņÜ®ņØä ļ¦żĻ░£ĒĢśļŖö ņŗĀĒśĖ ļČäņ×ÉļĪ£, Ļ│äņŖ╣ņ╣śĻ░Ć ļ░£ļŗ¼ĒĢśļŖö ņ£äņ╣śņŚÉņä£ ņ╣śņĢä ņāüĒö╝ņØś ņ”ØņŗØņØä ņ×ÉĻĘ╣ĒĢśĻ│Ā, ņāüĒö╝ņØś ņä▒ņןņØä ļÅĢļŖö ņŚŁĒĢĀņØä ĒĢ£ļŗż[20]. ShhĻ░Ć Ļ│╝ļ░£ĒśäļÉśļ®┤ ņāüĒö╝ ļÅīĻĖ░Ļ░Ć ņäżņĖĪņ£╝ļĪ£ ņŚ░ņןļÉśĻ│Ā, ņČöĻ░ĆņĀüņØĖ ņ╣śņŚ┤ņØś ĒśĢņä▒ņØä ņ£ĀļÅäĒĢĀ ņłś ņ׳ļŗż. ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ļÅä ņ╣śĒīÉņØ┤ ļČłņÖäņĀäĒĢśĻ▓ī ĒØĪņłśļÉśļŖö Ļ▓āņØ┤ Ļ┤Ćņ░░ļÉśĻ│Ā, ņØ┤ļŖö RUNX2ņØś Ļ░ÉņåīļĪ£ ņØĖĒĢ┤ ņśüĻĄ¼ņ╣śņŚ┤ ņÖäņä▒ ņØ┤ĒøäņŚÉļÅä ņ╣śĒīÉņØ┤ ņ×öņĪ┤ĒĢśņŚ¼ Ļ│╝ņ×ēņ╣śņØś ĒśĢņä▒ņŚÉ ĻĖ░ņŚ¼ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņ£ĀņČöĒĢĀ ņłś ņ׳ļŗż[21].
ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×ÉņŚÉņä£ ļéśĒāĆļéśļŖö ņØ┤ļ¤¼ĒĢ£ ņ╣śņä▒ ļ¼ĖņĀ£ļŖö ņä▒ņןĻ│╝ņĀĢņŚÉņä£ ņĪ░ĻĖ░ņŚÉ Ļ░£ņ×ģĒĢśņŚ¼ ĒĢ┤Ļ▓░ĒĢśļŖö Ļ▓āņØ┤ ņ£Āļ”¼ĒĢśļŗż[22]. ļ¦╣ņČ£ ņ¦ĆņŚ░ņØ┤ ņśłņāüļÉśļŖö ņśüĻĄ¼ņ╣śņØś ņ╣śĻĘ╝ņØ┤ 1/3ņĀĢļÅä ņ×Éļ×ÉņØä ļĢī, ĒĢ┤ļŗ╣ ļČĆņ£äņŚÉ ņĪ┤ņ×¼ĒĢśļŖö Ļ│╝ņ×ēņ╣śņÖĆ, ņāüļ░®ņØś Ļ│© ļ░Å ņ£Āņ╣śļź╝ ĒĢ©Ļ╗ś ņĀ£Ļ▒░ĒĢ©ņ£╝ļĪ£ņŹ© ņśüĻĄ¼ņ╣śņØś ņ×Éļ░£ņĀüņØĖ ļ¦╣ņČ£ņØä ĻĖ░ļīĆĒĢĀ ņłś ņ׳ļŗż. ļśÉĒĢ£ Ļ│╝ņ×ēņ╣śĻ░Ć Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖļŖö Ļ▓ĮņÜ░ņŚÉļÅä, ņśüĻĄ¼ņ╣śņØś ņ╣śĻĘ╝ņØ┤ 1/2ņĀĢļÅä ņä▒ņןĒ¢łņØä ļĢī, ļ¦żļ│ĄļÉ£ ņśüĻĄ¼ņ╣ś ņāüļ░®ņØś Ļ│©Ļ│╝ ņ£Āņ╣śļź╝ ņĀ£Ļ▒░ĒĢśĻ│Ā ņÖĖĻ│╝ņĀü ļģĖņČ£ņØä ļÅÖļ░śĒĢ©ņ£╝ļĪ£ņŹ© ņśüĻĄ¼ņ╣śņØś ļ¦╣ņČ£ņØä ļÅäļ¬©ĒĢśĻ│Ā ĻĄÉņĀĢņĀü ņ╣śļŻīĻĖ░Ļ░äņØä ļŗ©ņČĢĒĢĀ ņłś ņ׳ļŗż.
ņØ┤ ņŚ░ĻĄ¼ļź╝ ĒåĄĒĢ┤ ļ░£Ļ▓¼ļÉ£ ļÅīņŚ░ļ│ĆņØ┤ļŖö RHD ņśüņŚŁ ļé┤ņŚÉņä£ ļ░£ņāØĒĢ£ ļÅīņŚ░ļ│ĆņØ┤ļĪ£, ļ│ĆņØ┤ļÉ£ mRNAĻ░Ć nonsense-mediated mRNA decay (NMD) systemņŚÉ ņØśĒĢ┤ ņĪ░ĻĖ░ņŚÉ ļČäĒĢ┤ļÉĀ ņłś ņ׳ļŗż. ļśÉļŖö, ņāłļĪŁĻ▓ī ĒśĢņä▒ļÉ£ ņĀĢņ¦Ć ņĮöļÅłņØ┤ ņĪ░ĻĖ░ ļ▓łņŚŁ ņóģĻ▓░ņØä ņ┤łļלĒĢśĻ│Ā, ļ│ĆĒśĢļÉ£ ļŗ©ļ░▒ņ¦łņØĆ DNA Ļ▓░ĒĢ® ĻĖ░ļŖźņØ┤ ņåīņŗżļÉśņ¢┤ ņĀäņé¼ ņØĖņ×ÉļĪ£ņä£ņØś ĒÖ£ņä▒ņØä ņ×āĻ▓ī ļÉĀ ņłś ņ׳ļŗż. Ļ▓░Ļ│╝ņĀüņ£╝ļĪ£ ņĀĢņāüņĀüņØĖ RUNX2ņØś ļ░£Ēśäņ¢æņØ┤ Ļ░ÉņåīĒĢśņŚ¼ ļŗ©ņāüļČĆņĪ▒(haploinsufficiency)ņØä ņ£Āļ░£ĒĢĀ ņłś ņ׳ļŗż.
ļŗ©ņāüļČĆņĪ▒ņØ┤ļ×Ć, ļ░░ņłśņ▓┤ ņāØļ¼╝ņŚÉņä£ ļ│ĆņØ┤ļĪ£ ņØĖĒĢ┤ ļ░śņłśņ▓┤Ļ░Ć ļČłĒÖ£ņä▒ĒÖöļÉśņ¢┤, ĻĖ░ļŖźņØä ĒĢĀ ņłś ņ׳ļŖö ņ£ĀņĀäņ×ÉņØś ņ¢æņØ┤ ļ░śņ£╝ļĪ£ Ļ░ÉņåīĒĢśņŚ¼ ņ¦łĒÖśņØ┤ ņ£Āļ░£ļÉśļŖö Ēśäņāüņ£╝ļĪ£, ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”ØņØĆ ļŗ©ņāüļČĆņĪ▒ņØ┤ ļéśĒāĆļéśļŖö ņ¦łĒÖś ņżæ ĒĢśļéśļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. Lou ļō▒[23]ņØĆ Runx2ņØś ĒŖ╣ņ¦ĢņĀüņØĖ ļ│ĆņØ┤ļź╝ ņØ┤ņÜ®ĒĢśņŚ¼, ņźÉņŚÉņä£ Runx2 mRNAņØś ļ░£Ēśäņ¢æņŚÉ ļö░ļźĖ Ēæ£ĒśäĒśĢņØś ļ░£Ēśä ņ¢æņāüņØä ĒÖĢņØĖĒĢśņśĆļŗż. Runx2 mRNAĻ░Ć ņĀĢņāüĻĄ░ņØś 70% ņØ┤ĒĢśļĪ£ ļ░£ĒśäļÉĀ ļĢī ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”ØņØś Ēæ£ĒśäĒśĢņØ┤ ļéśĒāĆļé¼Ļ│Ā, ņĀĢņāüņĀüņØĖ Ļ│©Ļ▓®ņØś ĒśĢņä▒ņØä ņ£äĒĢ┤ņä£ļŖö 79%ņØ┤ņāü ļ░£ĒśäļÉśņ¢┤ņĢ╝ ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłļŗż. ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£, ņØ┤ ņŚ░ĻĄ¼ņŚÉņä£ ĒÖĢņØĖļÉ£ RUNX2ņØś ļÅī ņŚ░ļ│ĆņØ┤ļĪ£ ņØĖĒĢ┤ RUNX2ņØś ļ░£Ēśäņ¢æņØ┤ ņČ®ļČäĒĢśņ¦Ć ļ¬╗ĒĢ┤ ļīĆņāüņ×ÉļōżņŚÉņä£ ņśüĻĄ¼ņ╣ś ļ¦╣ņČ£ ņ¦ĆņŚ░ ļō▒ņØ┤ ļéśĒāĆļé¼ņØä Ļ░ĆļŖźņä▒ņØ┤ ļåÆļŗż.
ĒĢ£ĒÄĖ, ņØ┤ ņŚ░ĻĄ¼ņØś ņ░ĖņŚ¼ ļīĆņāüņ×ÉļōżņŚÉņä£ ņśüĻĄ¼ņ╣ś ļ¦╣ņČ£ ņ¦ĆņŚ░ņØĆ ļéśĒāĆļé¼ņ¦Ćļ¦ī ĒŖ╣ņØ┤ņĀüņ£╝ļĪ£ Ļ│╝ņ×ēņ╣śļŖö Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖņĢśļŗż. ļŗ©ņāüļČĆņĪ▒ņŚÉ ņØśĒĢ┤ ļéśĒāĆļéśļŖö ņ¦łĒÖśņŚÉņä£, Ēæ£ĒśäĒśĢņØĆ ļŗżņ¢æĒĢ£ ņ£ĀņĀäņĀü ņłśņĀĢ ņÜöņØĖ(modifier) Ēś╣ņØĆ ĒÖśĻ▓Į ņÜöņØĖņØś ņśüĒ¢źņØä ļ░øĻ▓ī ļÉśĻ│Ā, ļŗżņ¢æĒĢ£ ņĪ░ņ¦üņŚÉņä£ ņāüņØ┤ĒĢ£ Ļ▓░Ļ│╝ļĪ£ ļéśĒāĆļé£ļŗż. ļÅÖņØ╝ĒĢ£ RUNX2 ļÅīņŚ░ļ│ĆņØ┤Ļ░Ć ĒÖĢņØĖļÉ£ ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Ø ĒÖśņ×É ņ”ØļĪĆļōżņŚÉņä£ Ļ│╝ņ×ēņ╣śņØś ņłśņÖĆ ņ£äņ╣śĻ░Ć ļŗżņ¢æĒĢśĻ▓ī ļ│┤Ļ│ĀļÉśņŚłļŗż[10,16,24]. ļö░ļØ╝ņä£, ņĢäņ¦üĻ╣īņ¦Ć RUNX2ņØś Ļ▓░ĒĢ© ļČĆņ£äņÖĆ Ļ│╝ņ×ēņ╣ś Ļ░äņØś ļ¬ģĒÖĢĒĢ£ ņāüĻ┤ĆĻ┤ĆĻ│äļź╝ Ļ▓░ņĀĢĒĢśĻĖ░ļŖö ņ¢┤ļĀĄĻ│Ā, RUNX2 ņØ┤ņÖĖņØś ļŗżņ¢æĒĢ£ ņÜöņåīļōżņØ┤ Ļ│╝ņ×ēņ╣śņØś ļ░£ņāØ Ļ│╝ņĀĢņŚÉ Ļ┤ĆņŚ¼ĒĢ£ļŗżĻ│Ā ļ│╝ ņłś ņ׳ļŗż.
ņØ┤ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ĒŖ╣ņ¦ĢņĀüņØĖ ĻĄ¼Ļ░Ģ ļé┤ Ēæ£ĒśäĒśĢņØä ņ¦ĆļŗłļŖö ņāłļĪ£ņÜ┤ RUNX2ņØś ļÅīņŚ░ļ│ĆņØ┤ļź╝ ĒÖĢņØĖĒĢśņśĆļŗż. RUNX2ņØś Ļ▓░ĒĢ© ļČĆņ£äņŚÉ ļīĆĒĢ£ ņØ┤ņĀäņØś ņŚ░ĻĄ¼ļōżĻ│╝ņØś ļ╣äĻĄÉņÖĆ ņ×äņāü ņ¢æņāüņØś ļ░£ĒśäĻĖ░ņĀäņŚÉ ļīĆĒĢ£ ņČöĻ░ĆņĀüņØĖ ņŚ░ĻĄ¼ļŖö ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”ØņØś ņ£ĀņĀäņ×ÉĒśĢĻ│╝ Ēæ£ĒśäĒśĢņØś Ļ┤ĆĻ│äļź╝ ĻĘ£ļ¬ģĒĢśļŖöļŹ░ ļÅäņøĆņØ┤ ļÉĀ Ļ▓āņØ┤ļŗż.
Ōģż. Ļ▓░ ļĪĀ
ņØ┤ ņŚ░ĻĄ¼ļź╝ ĒåĄĒĢ┤ ņćäĻ│© ļæÉĻ░£ ņØ┤ĒśĢņä▒ņ”Øņ£╝ļĪ£ ņ¦äļŗ©ļÉ£ ļīĆņāüņ×ÉņŚÉņä£ RUNX2 ņ£ĀņĀäņ×ÉņØś ņāłļĪ£ņÜ┤ ļÅīņŚ░ļ│ĆņØ┤[NM_001024630.4: c.357delC, p.(Asn120Thrfs*24)]ļź╝ ļ░£Ļ▓¼ĒĢśņśĆļŗż. ĒĢ┤ļŗ╣ ļÅīņŚ░ļ│ĆņØ┤ļŖö RUNX2ņØś ļŗ©ņāüļČĆņĪ▒ņØä ņ┤łļלĒĢśņŚ¼, ņ╣śņĢä ļ░Å ņŻ╝ņ£ä ņĪ░ņ¦üņØś ļ░£ņāØĒĢÖņĀü Ļ▓░ĒĢ©ņØä ņĢ╝ĻĖ░ĒĢśĻ│Ā, ĒĢ┤ļŗ╣ Ļ░ĆĻ│äņŚÉņä£ ļéśĒāĆļé£ ņśüĻĄ¼ņ╣ś ļ¦╣ņČ£ ņ¦ĆņŚ░ņØś ļ│æņØĖņ£╝ļĪ£ ņ×æņÜ®ĒĢśņśĆņØä Ļ▓āņ£╝ļĪ£ ņé¼ļŻīļÉ£ļŗż. ņØ┤ļŖö ņØ┤ņĀäņØś RUNX2 ņ£ĀņĀäņ×ÉņŚÉņä£ ļ░ØĒśĆņ¦ä ļŗżņ¢æĒĢ£ ļÅīņŚ░ļ│ĆņØ┤ņÖĆ ĒĢ©Ļ╗ś, Ē¢źĒøä ĒĢ┤ļŗ╣ ņ£ĀņĀäņ×ÉņØś ļÅīņŚ░ļ│ĆņØ┤ņÖĆ ņØ┤ļĪ£ ņØĖĒĢ£ Ēæ£ĒśäĒśĢņØś Ļ┤ĆĻ│äļź╝ ņäżļ¬ģĒĢśļŖöļŹ░ ļÅäņøĆņØ┤ ļÉĀ Ļ▓āņØ┤ļŗż.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print